
文件管理是質(zhì)量保證系統(tǒng)的基本要素,為影響藥品質(zhì)量的活動提供了可追溯、可驗證的書面證據(jù)。PIC/S GMP第四章與中國GMP第八章均對文件管理提出了詳盡要求。兩者在基本原則和核心要求上高度一致,覆蓋范圍基本包括質(zhì)量標(biāo)準(zhǔn)、工藝規(guī)程/說明、批記錄、操作規(guī)程和記錄等。
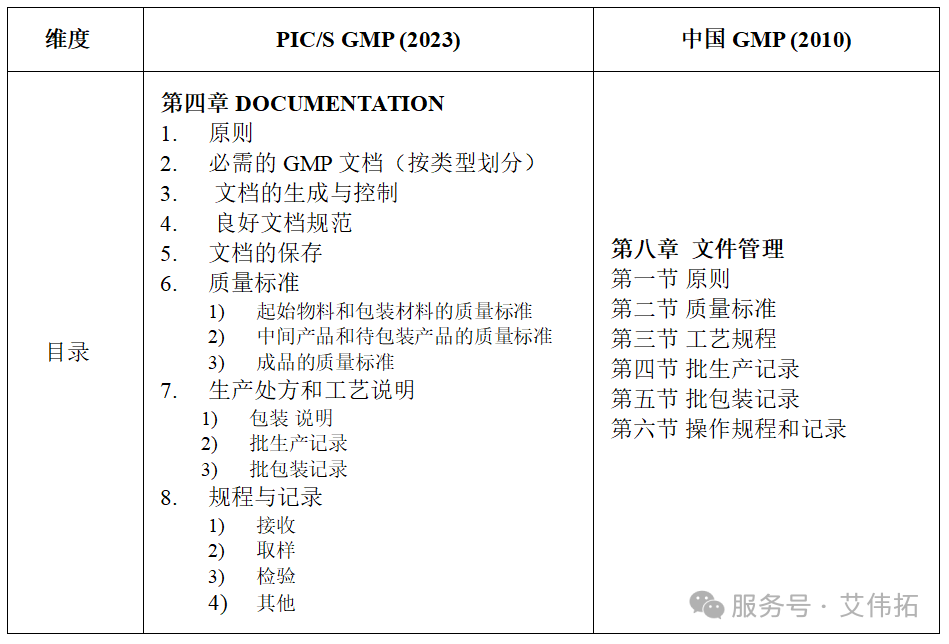
質(zhì)量標(biāo)準(zhǔn)
兩者均明確要求為物料、成品應(yīng)當(dāng)有經(jīng)批準(zhǔn)的質(zhì)量標(biāo)準(zhǔn),其要求包含的核心內(nèi)容基本一致。對中間產(chǎn)品,兩者都規(guī)定,在中間產(chǎn)品被外購/外銷或其質(zhì)量評價與成品質(zhì)量直接相關(guān)時,應(yīng)當(dāng)制定其質(zhì)量標(biāo)準(zhǔn)。在效期管理方面,PIC/S GMP默認(rèn)物料通常設(shè)定“the maximum period of storage before re-examination"。中國GMP則為物料提供了有效期和復(fù)驗期兩種選項。
工藝規(guī)程
每種產(chǎn)品的每個生產(chǎn)批量均應(yīng)當(dāng)有經(jīng)企業(yè)批準(zhǔn)的工藝規(guī)程。生產(chǎn)處方均要求明確產(chǎn)品信息、物料清單和產(chǎn)量說明。生產(chǎn)操作說明都涵蓋生產(chǎn)場所、設(shè)備、步驟、控制和貯存。包裝操作要求兩者均涵蓋包裝規(guī)格/形式、包裝材料清單、印刷包材樣稿、清場要求、操作步驟描述等基本要素。
批生產(chǎn)記錄
兩者GMP均強調(diào)批生產(chǎn)記錄必須基于現(xiàn)行批準(zhǔn)的工藝文件,兩者關(guān)鍵內(nèi)容絕大部分是重合的。中國 GMP 的定更為全面和細(xì)致,它不僅規(guī)定了記錄應(yīng)包含的內(nèi)容(中國GMP第175條),還延伸至記錄本身,例如要求“避免填寫差錯",批生產(chǎn)記錄的每一頁應(yīng)當(dāng)標(biāo)注產(chǎn)品的名稱、規(guī)格和批號(第172條);原版空白記錄需經(jīng)生產(chǎn)和質(zhì)量負(fù)責(zé)人共同批準(zhǔn),且每批產(chǎn)品的生產(chǎn)只能發(fā)放一份原版空白批生產(chǎn)記錄的復(fù)制件(第173條)。
批包裝記錄
中國GMP 第五節(jié) 批包裝記錄總結(jié)了包裝環(huán)節(jié)的文檔管理要求。它規(guī)定每批產(chǎn)品包裝均需建立可追溯的批包裝記錄(第176條),記錄需依據(jù)工藝規(guī)程制定(第177條),內(nèi)容涵蓋產(chǎn)品信息、包裝材料、包裝操作、物料平衡檢查及異常處理等內(nèi)容(第180條)。包裝操作需實時記錄并由操作人員簽字確認(rèn)(第179條)。PIC/S 批包裝記錄內(nèi)容與中國GMP總體相似,其條款4.21 (h),所有印刷包裝材料及待包裝產(chǎn)品的名稱、代碼,以及發(fā)放、使用、銷毀或退庫的數(shù)量等,均應(yīng)記錄以便進(jìn)行充分核對。若包裝過程中已實施可靠的電子控制措施,則可能需要不包含此信息的理由。
操作規(guī)程和記錄
中國GMP僅三條內(nèi)容(第181條至第183條),規(guī)定了操作規(guī)程應(yīng)包含的要素,并要求對廠房、設(shè)備、物料、文件和記錄等實施惟一性編碼,同時對確認(rèn)、驗證、偏差處理等關(guān)鍵活動也需建立相應(yīng)規(guī)程并記錄。PIC/S GMP補充了物料接收、取樣、檢驗等環(huán)節(jié)更細(xì)致的記錄要求。
參考依據(jù):
【1】 PIC/S: Guide to GMP for Medicinal Products part Ⅰ (PE 009-17, August 2023) Ⅰ部
【2】 《藥品生產(chǎn)質(zhì)量管理規(guī)范(2010年修訂)》
消息")
